近日,在美國化學會(hui) (ACS)2024春季會(hui) 議的“首次公開(First-time disclosures)”環節中,六家生物醫藥公司首次披露了他們(men) 正在研發中的抗腫瘤小分子藥物的化學結構和初期臨(lin) 床試驗數據。這些潛在的新藥針對的靶點包括免疫檢查點蛋白和多個(ge) 與(yu) 癌症進展密切相關(guan) 的關(guan) 鍵酶,這些靶點由於(yu) 多種原因難於(yu) 靶向。這六款小分子的結構設計和作用機製上呈現出創新性,也展現了業(ye) 界在攻克難於(yu) 靶向蛋白靶點時的多種解決(jue) 方案。藥明康德內(nei) 容團隊將依據ACS旗下C&EN網站的報道及公開信息,向讀者詳細介紹這些創新分子結構及其相應的臨(lin) 床進展。
候選藥物:ORIC-533
研發機構:Oric Pharmaceuticals
靶點:CD73
疾病領域:多發性骨髓瘤
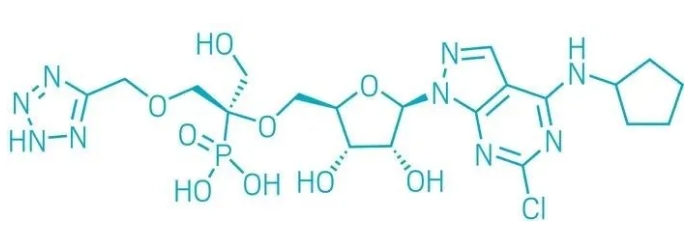
▲ORIC-533的結構式(圖片來源:參考資料[1])
ORIC-533是一種抑製腺苷生成過程中的關(guan) 鍵酶CD73的口服候選藥物。CD73是一種控製細胞外腺苷產(chan) 生速率的酶,它的過度活躍與(yu) 多種癌症的不良預後有關(guan) 。該酶是免疫抑製途徑的一部分,與(yu) 多發性骨髓瘤和其他癌症的耐藥性相關(guan) 。開發這類藥物的挑戰在於(yu) ,即使在高濃度的單磷酸腺苷(CD73的常規底物)競爭(zheng) 性地與(yu) CD73結合的情況下,開發的藥物分子仍能保持藥效。
ORIC-533分子的核心是腺苷衍生物,研究人員通過增強氫鍵形成並且同時平衡整體(ti) 電荷和極性,以保持較高的生物利用度。他們(men) 發現,膦酸酯基團周圍的區域對於(yu) 在生化試驗中阻斷腺苷產(chan) 生的效果影響最大,該基團在蛋白質和鋅離子之間形成鹽橋。此外,通過醚鍵連接的四唑與(yu) CD73活性位點中的三個(ge) 關(guan) 鍵氨基酸形成氫鍵,從(cong) 而增強了候選藥物的功效。
目前,ORIC-533正在針對多發性骨髓瘤患者開展1b期臨(lin) 床試驗,它是三種正處於(yu) 臨(lin) 床試驗階段的CD73小分子抑製劑之一。初步的臨(lin) 床試驗數據顯示,ORIC-533具有良好的藥代動力學特征,對可溶性CD73酶活性具有強效的抑製作用,顯示出了良好的靶向作用。該藥物總體(ti) 耐受性良好,主要觀察到的治療相關(guan) 不良事件為(wei) 1級和2級,沒有發現任何特定的複發性毒性、劑量限製性毒性、劑量減少的情況或與(yu) 治療相關(guan) 的嚴(yan) 重不良事件。
目前,Oric Pharmaceuticals正計劃與(yu) 輝瑞(Pfizer)合作開展2期臨(lin) 床研究。
候選藥物:NX-1607
研發機構:Nurix Therapeutics
靶點:CBL-B(Casitas B細胞淋巴瘤蛋白B)
疾病領域:癌症
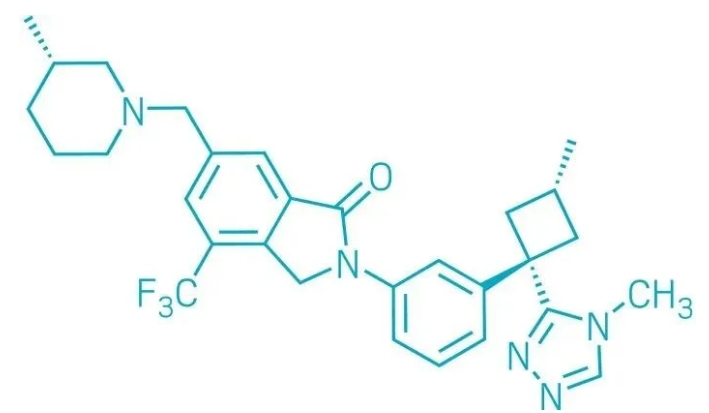
▲NX-1607的結構式(圖片來源:參考資料[1])
Nurix Therapeutics開發的NX-1607是一種靶向CBL-B的創新免疫療法。CBL-B作為(wei) 一種E3泛素連接酶,在多種免疫細胞中發揮作用,通過促進關(guan) 鍵下遊信號蛋白(如Vav1,PLCγ)的泛素化和降解,對免疫功能進行負向調節。在CBLB基因敲除的小鼠模型中,觀察到了對移植腫瘤的抵抗性,展示了CBL-B作為(wei) 一種新型細胞內(nei) 免疫檢查點的潛力。然而,由於(yu) CBL-B缺乏明確的小分子對接位點,使其成為(wei) 一個(ge) 具有挑戰性的藥物靶點。
NX-1607的獨特之處在於(yu) 其作為(wei) 一種分子膠,能夠與(yu) CBL-B結合並促使其與(yu) 不同亞(ya) 基結合,從(cong) 而將CBL-B鎖定在非活性構象中,阻止其向活性狀態轉換。這種機製有效地抑製了CBL-B的功能。通過高通量篩選技術,研究團隊發現,將N-甲基三唑結構引入至候選分子中,能夠有效地填補蛋白質的結合口袋,其中三唑的硝基通過氫鍵與(yu) 蛋白質形成穩定結合,延長了分子與(yu) 蛋白的結合時間。環丁烷基團作為(wei) 間隔結構(spacer),有助於(yu) 確保三唑以正確的角度進入結合口袋,而一個(ge) 苄胺基團則插入到蛋白的另一個(ge) 關(guan) 鍵疏水口袋結構中。
在臨(lin) 床前研究中,NX-1607顯著提升了T細胞的活性,並在單藥治療以及與(yu) 抗PD-1免疫檢查點抑製劑聯合使用時展現出抗腫瘤效果。得益於(yu) 其良好的溶解性和高滲透性,NX-1607的生產(chan) 過程相對簡便。目前,針對晚期實體(ti) 瘤患者的NX-1607單藥以及與(yu) 紫杉醇聯合使用的1期臨(lin) 床試驗正在進行中。
候選藥物:SGR-1505
研發機構:Schrödinger
靶點:MALT1
疾病領域:B細胞淋巴瘤
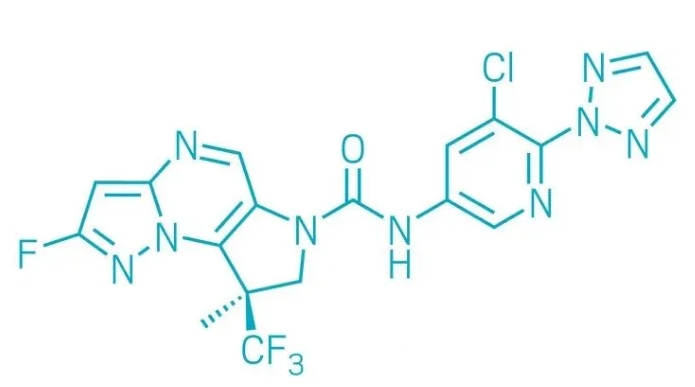
▲SGR-1505的結構式(圖片來源:參考資料[1])
SGR-1505是Schrödinger公司利用其專(zhuan) 有基於(yu) 物理原理的計算平台發現的一種MALT1蛋白靶向療法。該療法的設計理念是通過與(yu) MALT1蛋白的一個(ge) 變構位點結合,從(cong) 而使癌細胞中的MALT1蛋白失活。
MALT1是一種位於(yu) 布魯頓氏酪氨酸激酶(BTK)下遊信號的蛋白酶,是多種非霍奇金B細胞淋巴瘤的潛在治療靶點。對於(yu) 那些對現有BTK抑製劑產(chan) 生抗藥性的癌症病患,MALT1提供了一個(ge) 有希望的備選靶點。
利用高級計算模型預測蛋白質與(yu) 小分子之間的相互作用,Schrödinger公司能夠從(cong) 數千個(ge) 候選結構中篩選出最具潛力的候選物,並將其送入實驗室進行製造和測試。這一藥物開發過程耗時約10個(ge) 月,經過對超過80億(yi) 個(ge) 化合物的計算評估,最終選出了129種分子進行合成。
目前,SGR-1505正在晚期B細胞淋巴瘤患者中進行1期臨(lin) 床試驗。Schrödinger的研究團隊預計,初步數據將於(yu) 2024年底或2025年公布,屆時將為(wei) B細胞淋巴瘤的治療提供新的見解。
候選藥物:DCC-3116
研發機構:Deciphera Pharmaceuticals
靶點:ULK1/2
疾病領域:與(yu) RAS/RAF基因突變有關(guan) 的癌症
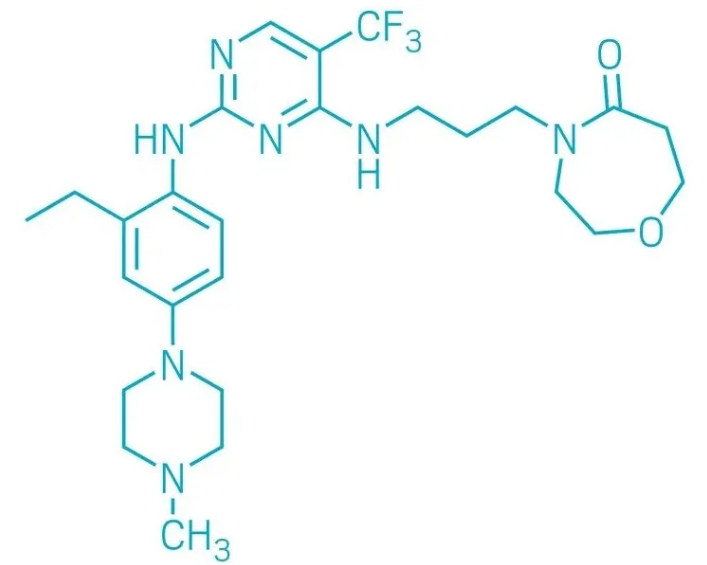
▲DCC-3116的結構式(圖片來源:參考資料[1])
在許多癌症類型中,受體(ti) 酪氨酸激酶(RTK)通路的突變非常普遍,因此,針對RTK蛋白的治療手段不斷湧現。但癌細胞能通過啟動自噬來抵禦這些治療手段,自噬通過回收和再利用損壞的蛋白質幫助癌細胞存活。ULK1和ULK2蛋白是自噬過程的關(guan) 鍵組分,Deciphera公司的研究團隊正致力於(yu) 開發一種抑製這兩(liang) 種蛋白的藥物,旨在阻斷癌細胞的自噬過程。
DCC-3116是Deciphera公司開發的一款選擇性的小分子ULK1/2激酶抑製劑,旨在通過抑製負責啟動自噬的ULK1/2激酶來抑製癌症自噬,從(cong) 而影響腫瘤細胞的存活。它擁有一個(ge) 中央嘧啶基團,並攜帶兩(liang) 個(ge) 仲胺基團形成的雙臂結構,這使得它能與(yu) 目標蛋白結合並將其鎖定在非活性構象中。
DCC-3116與(yu) 其他具有相似結構的候選藥物相比,具有更高的藥效和更緩慢的解離速度。這一特性歸因於(yu) 其分子結構中的兩(liang) 臂與(yu) 蛋白質之間的優(you) 化相互作用:甲基呱嗪與(yu) 蛋白中的關(guan) 鍵天冬氨酸殘基發生相互作用,同時,內(nei) 酰胺環恰好適配於(yu) 酶的P環形成的口袋結構。
在動物模型中,DCC-3116展現了有效的自噬抑製和抗腫瘤活性。基於(yu) 這些前期成果,Deciphera公司目前正在進行DCC-3116的單藥治療以及與(yu) RTK抑製劑聯合使用的1期臨(lin) 床試驗。Deciphera公司希望在今年晚些時候能夠確定用於(yu) 2期臨(lin) 床試驗的給藥劑量。
候選藥物:TNG348
研發機構:Tango Therapeutics
靶點:USP1
疾病領域:BRCA1/2突變癌症和其他同源重組缺陷(HRD+)癌症
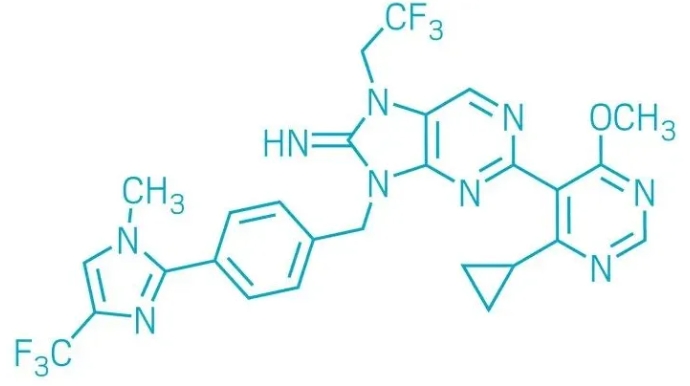
▲TNG348的結構式(圖片來源:參考資料[1])
TNG348是一種針對USP1的可逆變構抑製劑,被認為(wei) 是治療HRD+癌症的潛在藥物。在HRD+癌症中,DNA修複機製遭到破壞,Tango公司的研究團隊提出,抑製DNA修複酶USP1是消滅這類癌細胞的一種有效策略。
USP1是一種去泛素化酶,對於(yu) BRCA1/2突變和其他HRD+癌細胞的生存和增殖至關(guan) 重要。在BRCA1/2突變的情況下,通過抑製USP1可以誘導所謂的“合成致死”。正常細胞有多種機製來修複受損DNA並防止細胞死亡,而BRCA1/2突變細胞則依賴於(yu) 轉座子合成和堿基切除修複。抑製USP1會(hui) 影響轉座子合成,從(cong) 而阻礙DNA複製,導致BRCA1/2突變癌細胞死亡。
根據既往有關(guan) USP1抑製劑的文獻報道,Tango公司的研發團隊確定胍衍生物是一條很有前景的候選路徑。通過篩選多種苄基連接子和芳香基團上的官能團,研究團隊進一步優(you) 化了分子結構。為(wei) 了降低分子核心的堿性並減少對心髒鉀通道的潛在不良作用,他們(men) 在候選分子上引入了氟化基團。當TNG348與(yu) USP1的變構位點結合時,會(hui) 破壞酶內(nei) 的氫鍵網絡,從(cong) 而抑製其活性。
臨(lin) 床前研究結果表明,TNG348單獨使用或是與(yu) PARP抑製劑一起使用都能有效縮小腫瘤。目前,TNG348針對HRD+乳腺癌和卵巢癌患者的1期臨(lin) 床試驗已正式啟動,去年年底已對首位受試者進行了給藥。
候選藥物:BLU-222
研發機構:Blueprint Medicines
靶點:細胞周期蛋白依賴性激酶2(CDK2)
疾病領域:激素受體(ti) 陽性(HR+)/人表皮生長因子受體(ti) 2陰性(HER2-)乳腺癌和其他CCNE1變異癌症
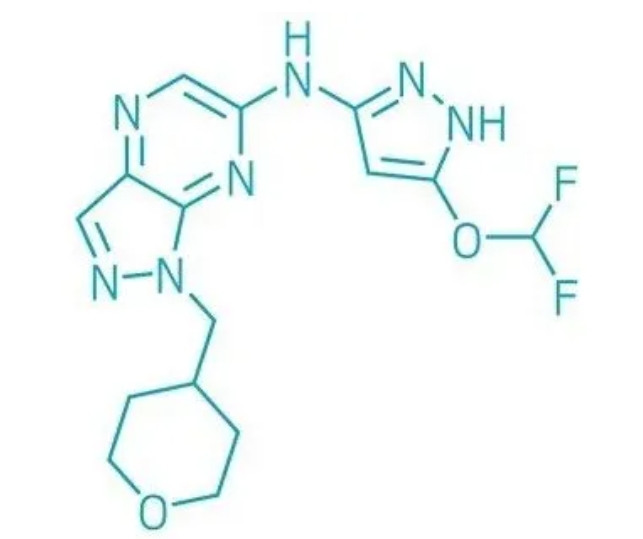
▲BLU-222的結構式(圖片來源:參考資料[1])
CDK是細胞分裂周期的關(guan) 鍵調節因素。其中,CDK2是一種非常有前景的藥物靶點,尤其是在那些Cyclin E1蛋白過度表達的癌症中,包括對CDK4/6抑製劑耐藥的HR+/HER2-乳腺癌。Blueprint公司的研發團隊專(zhuan) 注於(yu) 開發針對CDK2的特異性抑製劑,這一過程頗具挑戰性,因為(wei) CDK2的活性位點與(yu) CDK1極為(wei) 相似。
BLU-222是Blueprint公司開發的CDK2特異性抑製劑。Blueprint公司的科研人員充分篩選了公司的化合物庫,這個(ge) 庫詳細記錄了每種化合物與(yu) 所有人類激酶的相互作用情況,以篩選出特異性的CDK2先導化合物。結果表明,含有吡嗪核心的化合物提供了最佳的選擇性和活性組合。帶有二氟甲氧基基團的吡唑環能與(yu) “門控開關(guan) (gatekeeper)”苯基丙氨酸殘基中的π電子產(chan) 生關(guan) 鍵的相互作用,從(cong) 而進一步提高了其對其他激酶的選擇性。這些努力最終促成了BLU-222分子的誕生,該分子對CDK2的選擇性比對CDK1高出90倍。
目前,BLU-222正在進行1期臨(lin) 床試驗,不僅(jin) 作為(wei) 單藥治療方案,也在與(yu) CDK4/6抑製劑的聯合用藥方案中進行測試。
隨著2024年美國化學學會(hui) 春季大會(hui) 的成功閉幕,一批候選抗癌藥物的亮相標誌著產(chan) 業(ye) 界在抗癌領域取得了更進一步的進展。期待與(yu) 這些候選新藥能夠獲得深入研究和進一步驗證,在不遠的將來為(wei) 癌症患者提供更為(wei) 優(you) 化和有效的治療選擇。